La glucólisis y el ciclo del ácido cítrico generan una cantidad relativamente baja de ATP. Sin embargo seis pasos de deshidrogénación (uno en la glucolisis, otro en la reacción de la piruvato deshidrogenada y cuatro más en el ciclo del ácido cítrico) reducen en total 10 moles de NAD+ a NADH2 y dos moles de FAD a FADH2 por mol de glucosa. La reoxidación de estos transportadores electrónicos reducidos genera la mayor parte de la energía necesaria para la síntesis de ATP.
Así es como se llega a una serie de procesos que tienen lugar en la respiración celular: el transporte electrónico (cadena respiratoria) y la fosforilación oxidativa.
Cadena respiratoria o cadena transportadora de electrones.
Se le llama cadena respiratoria al grupo de enzimas mitocondriales, acopladas, que actúan en estrecho contacto físico para catalizar este transporte electrónico. De esta forma, el nombre de cadena transportadora de electrones se refiere a la definición de oxidación y reducción, como pérdida o ganancia de electrones. El término cadena respiratoria apunta a que las reacciones acopladas implican un consumo de O2, es decir, respiración.
Como se mencionó anteriormente, los productos primarios de las reacciones del ciclo del ácido tricarboxílico son las coenzimas reducidas FADH2 y NADH, que contienen los electrones tomados de los diferentes sustratos oxidados.
Se llega a un punto importante, las mitocondrias no tienen la capacidad de impulsar el NADH formado en el citoplasma mediante la glucolisis. En vez de eso, los electrones de NADH se emplean para reducir de bajo peso molecular, que entonces: 1) Puede entrar a las mitocondrias a través de una vía alterna denominada “vía alterna o lanzadera entre malato y aspartato” y reducir NAD a NADH, o 2) Transferir sus electrones al FAD, a través de una vía denominada “vía alterna entre glicerol y fosfato”, para producir FADH2.
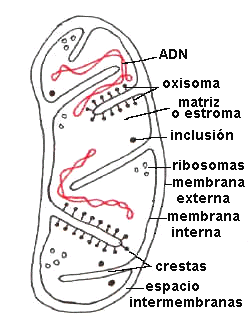
En la mitocondria donde los átomos de hidrógeno son sustraídos de los diversos sustratos por la acción de las deshidrógenasas; estas donan sus electrones a la cadena de transporte de electrones, la cual las transfiere al oxígeno molecular para producir agua. La energía liberada en el transporte de electrones se utiliza para la fosforilación oxidativa de ADP a ATP, proceso que se lleva a cabo en la mitocondria, es la membrana mitocondrial la que contiene la cadena transportadora de electrones y la ATP sintetasa, que es la mediadora de la formación de ATP.
La succinato deshidrogenasa del ciclo de Krebs, que forma parte de la cadena transportadora de electrones, es una proteína integral de la membrana interna mitocondrial.

Los componentes que intervienen en el transporte de electrones se han clasificado en complejos, denominados I, II, III y IV.
El complejo I esta representado por NADH: ubiquinona oxidorreductasa, y consta de 25 polipeptidos diferentes, además de flavina mononucléotido (FMN), complejos hierro azufre, ubiquinona o CoQ y fosofolípidos. Este complejo conocido como: (NADH-CoQ reductasa), cataliza la transferencia entre el NADH y la CoQ.
El complejo, al cual se acopla la síntesis de ATP en el sitio de acoplamiento 1 posee centros de hierro-azufre y un FMN. Este complejo es inhibido por el amital y la rotenona. Posee también el sitio de fosforilación 1.
El complejo II (succinato-CoQ reductasa) cataliza la reducción de la CoQ por los electrones removidos del succinato.
Este complejo que contiene FAD, está formado por cuantro polipéptidos con peso molecular de 70000, 27000, 15000 y 13000 D. Los dos polipéptidos mas grandes contienen el sitio catalítico para la oxidación del succinato; la subunidad mas pequeña (con peso molecular de 27000 D) de estos, dos péptidos contiene un centro de Fe4S4. Los otros dos péptidos de menor tamaño (con peso molecular de 15000 y 13000 D) poseen un centro de Fe2S2 y un citocromo tipo b, cuya función no se conoce.
El complejo III contiene dos citocromos tipo b, b560 y b562; un citocromo tipo c, c1; y un centro hierro-azufre. Posee también el sitio de fosforilación 2 y produce ATP conforme los electrones fluyen a través del complejo.
Este complejo es inhibido por el antibiótico antimicina.
En el complejo IV (citocromo c oxidasa), la citocromo oxidasa se ha reconocido desde hace mucho tiempo como la oxidasa Terminal de todas las células aeróbias, cataliza la oxidación por el O2 molecular del citocromo c reducido.
También se produce ATP conforme los electrones fluyen a través de la citocromo oxidasa. El complejo contiene citocromo a, dos iones de Cu2+ y citocromo a3.
El complejo IV es inhibido por el ion cianuro, el monóxido de carbono y la azida de sodio.
Secuencia de la cadena respiratoria.
Existen dos entradas o canales por los que los sustratos ceden los electrones a la cadena. El primer canal, denominado "sustratos NAD” , que es el mayoritario, en el están acopladas las deshidrogenasa correspondientes al piruvato, 2-oxoglutarato, malato, glutamato, isocitrato y 3-hidroxibutirato. Al segundo canal estan acopladas las deshidrogenasas del succinato, glicerol-fosfato y acil-CoA. Este ultimo se ha denominado canal de succinato y de manera mas general “de sustratos FAD”. Ambos canales confluyen en la coenzima Q, donde finaliza el transporte de átomos de hidrógeno, continuando el de electrones a través de los diferentes citocromos.

El complejo de la citocromo-oxidasa es el único miembro de la cadena mitocondrial de transporte de electrones que funcionan fisiológicamente con el oxigeno. Los radicales de oxígeno son productos secundarios naturales de la fosforilación oxidativa y corresponden aproximadamente al 1% del consumo total de oxígeno. Las fuentes principales de generación de radicales libres están en el complejo I y la CoQ y entre la CoQ y el complejo III. En estos sitios las Malvinas y las quinonas reducidas reaccionan con el oxígeno para producir varios radicales de oxígeno. Los radicales libres de superóxido, peróxido e hidroxilo reaccionan con proteínas y ácidos nucleicos y son toxico para la célula.
En el transporte electrónico mitocondrial los electrones se transfieren en formas diferentes, ya sea como iones hidruro o como átomos de hidrógeno, y en las ultimas etapas, catalizadas por los citocromos, como electrones.
Fosforilación oxidativa.
La síntesis de ATP se consigue por medio de un ensamblaje molecular situado en la membrana interna de la mitocondria. Este complejo enzimatico se denomina ATP sintetasa.
Los sitios de fosforilación son aquellos lugares de la cadena respiratoria donde se supone que la síntesis de ATP esta acoplada al transporte electrónico. Para la localización de estos se pueden utilizar criterios parecidos a los seguidos para la determinación de la secuencia de la cadena respiratoria. Puesto que el complejo IV es físicamente distinto de las proteínas que median el transporte electrónico, la energía libre liberada por el transporte electronico debe conservarse en una forma que pueda ser utilizada por la ATP sintetasa. Esta conservación de energía se conoce como; acoplamiento de energía o transducción de energía.

Hipótesis quimiosmótica.
Meter Mitchell en 1961, sugirió que el transporte de electrones y la síntesis de ATP estaban acopladas mediante un gradiente de protones a través de la membrana interna mitocondrial. Según este modelo, la transferencia de electrones a través de la cadena respiratoria produce un “bombeo de protones” desde la matriz mitocondrial al lado opuesto de la membrana interna de la mitocondria. En el lado citosolico aumenta la concentración de H+ y al mismo tiempo se genera un potencial electrico que en este lado de la membrana resulta positivo.
En esencia, la formación de una fuerza protomotriz, “bombeo de protones”, a través de la membrana interna mitocondrial inducida por el transporte de electrones, es el acontecimiento primario en la conservación de energía.
El modelo que propone Mitchell, puede resumirse en los siguientes postulado
1.-La membrana interna de la mitocondria es impermeable a los protones.
2.-la cadena transportadora de electrones está situada en la membrana interna de la mitocondia, de tal manera que mediante su funcionamiento asimétrico provoca la expulsión de protones.
3.-La ATPasa, sistema enzimático encargado de la síntesis de ATP, funciona asimétricamente, captando protones del exterior.
4.-Existe un sistema de difusión de cambio, probablemente H+/K+, que tiene por objeto disipar el gradiente de pH, sin elijar el potencial de membrana.
Consumo de oxígeno
La razón ADP/O se obtiene dividiendo la cantidad total de nanomoles de ADP adicionados entre los nanoátomos totales de oxígeno consumidos. Para sustratos que producen NADH el valor teórico es de 3, lo que quiere decir que cada átomo de oxígeno hasta que se consume se sintetizan 3 moléculas de ATP, para sustratos que reducen FADH2 el valor teórico de la ADP/O es de 2 moléculas de ATP. Calculos recientes atribuyen al NADH sólo 2.5 ATPs y al FADH2, sólo 1.5, por lo que una vuelta completa del ciclo tricarboxílico produciría, en vez de 12, solo 10 ATPs.
Genoma mitocondrial y enfermedades relacionadas.
Las mitocondrias son orgánulos semiautonomos que mantienen una relación endosimbiótica con la célula huésped. Estos orgánulos tienen su propio ADN, que codifica una serie de proteínas distintas y RNAs. El tamaño del genoma mitocondrial varía mucho según las especies.Las células que mitocondrias dependen de estos orgánulos para realizar la fosforilación oxidativa y, a cambio, la existencia misma de las mitocondrias depende de la célula.
Un evento endosimbiótico pudo haber tenido lugar en el momento en que un organismo libre capaz de realizar la fosforilación oxidativa fue engullido por otra célula. La doble membrana, el DNA circular (con algunas excepciones) y las maquinarias de transcripción y traducción especificas de las mitocondrias apuntan en este sentido. Gracias a la rápida acumulación de datos sobre la secuencia de genomas mitocondriales y bacterianos se puede especular con gran autoridad sobre el origen de la mitocondria “primigenia”. El genoma bacteriano que mas se parece al de las mitocondrias es el de Rickettsia prowazekii, el agente causante del tifus provocado por piojos. Los estudios sobre su secuencia sugieren que todas las mitocondrias existentes derivan de un ancestro de R. prowazekii y son el resultado de un único evento endosimbiótico.
La evidencia de que las mitocondrias actuales son el resultado de un único evento proviene del estudio del genoma mitocondrial más parecido al de las bacterias, el del protozoo Reclinomonas americana. Su genoma contiene 97 genes, de los cuales 62 codifican proteínas que representan la totalidad de los genes que codifican proteínas encontrados en todos los genomas mitocondriales encontrados.
Hay que tener en cuenta que en el mundo microbiano no es nada raro que una célula grande engulla de una forma transitoria células procarioticas. En lo que respecta a las mitocondrias esta relación transitoria se convirtió en permanente cuando la célula bacteriana perdió su DNA y su capacidad para vivir de forma independiente y la célula huésped paso a depender del ATP generado por su inquilino.
Como corresponde a un orgánulo tan importante para el metabolismo energético, el funcionamiento anomalo de las mitocondrias puede dar lugar a situaciones patológicas. El número de enfermedades que se pueden atribuir a mutaciones mitocondriales esta aumentando continuamente, al mismo ritmo que los conocimientos sobre la bioquímica y la genética de la mitocondrias.
La primera enfermedad mitocondrial conocida fue la neuropatía óptica hereditaria de Leber (NOHL), un tipo de ceguera que se produce en la madurez como resultado de las mutaciones en el componente NADH-Q oxidorreductasa del complejo I. Algunas de estas mutaciones impiden la utilización del NADH, mientras que otras bloquean la transferencia de electrones hasta Q. La acumulación de mutaciones en los genes mitocondriales a lo largo de varias décadas puede contribuir al envejecimiento, enfermedades degenerativas y cáncer.
El óvulo humano alberga varíos cientos de miles de moléculas de DNA mitocondrial, mientras el espermatozoide aporta unicamente unos pocos centenares, de modo que afecta muy poco al genotipo mitocondrial. Dado que el número de mitocondrias heredadas de la madre es muy grande y que no todas las mitocondrias se pueden ver afectadas, las patológias asociadas a mutaciones mitocondriales pueden ser bastante complejas. Incluso dentro de una única familia portadora de una misma mutación, las fluctuaciones al azar del porcentaje de mitocondrias mutadas dan lugar a una gran diversidad tanto en la naturaleza y gravedad de los síntomas asociados a la patología como en el periodo de tiempo en el que aparecen. A medida que aumenta el número de mitocondrias defectuosas disminuye la capacidad de generar energía hasta llegar a un punto en el cual la célula ya no puede funcionar correctamente. Los fallos en la respiración celular son peligrosos por partida doble. No solo disminuye la transducción de enegía sino que aumenta la probabilidad de que se generen derivados reactivos del oxígeno. Los orgános que dependen esencialmente de la fosforilación oxidativa, como el sistema nervioso y el corazón, son los mas sensibles a mutaciones del DNA mitocondrial.
Las mitocondrias desempeñan un papel clave en la apoptosis.
En el transcurso del desarrollo o en casos de graves daños celulares, células individuales pertenecientes a un organismo multicelular pueden experimentar la muerte celular programada o apoptosis. Las mitocondrias funcionan cono centros de control que regulan este proceso. Aunque todavía no se han establecido los detalles, en las mitocondrias dañadas se forma un poro que recibe el nombre de poro de transición de la permeabilidad de las mitocondrias (mtPTP). Aparentemente, este poro esta formado por VDAC (el translocador de nucleótidos de adenina) y algunas otras proteínas mitocondrailes, entre las que se incluyen miembros de una familia (familia Bcl) que fue descubierta, en principio, en su aparición en el cáncer. Uno de los activadores mas potentes de la apoptosis es el citocromo c. Su presencia en el citosol activa toda una cascada de enzimas proteolíticas llamadas caspasas. Estas cisteinproteasas se han conservado a lo largo de la evolución y se han encontrado en organismos que van desde la hidra hasta los seres humanos. El citocromo c, junto con otras proteínas, inicia la cascada mediante la activación de la procaspasa 9 a caspasa 9 que posteriormente activa otras caspasas. La activación de la cascada de caspasas, no provoca la destrucción generalizada de proteínas. Por el contrario, las caspasas tienen objetivos muy concretos. Por ejemplo, se destruyen las proteínas que mantienen la estructura de la célula. Otro ejemplo de la degradación de una proteína que inhibe a una enzima que destruye el DNA (Danza activada por caspasa, CAD) dejando el paso libre para que la CAD destruya el material genético. Esta cascada de enzimas proteolíticas recibe el nombre de “muerte por multitud de pequeños cortes”.
No hay comentarios:
Publicar un comentario